What next? - Utilization of transcriptome pro filing, proteome profiling and other taxonomic studies
Of course, DNA data are not an almighty means for understanding the life of plants, even if we limit our interests to the taxonomic treatment of a particular plant.
One example may be in examples of polyploidy and aneuploidy. Populations of Cayratia japonica (Thunb.) Gagn. (Vitaceae) occur as diploids and triploids (Okada et al., 2003). Our preliminary analysis of the trnL-F region of the chloroplast DNA did not detect any polymorphisms that could distinguish diploid individuals from triploid individuals, although the two types are two separate genetic populations. Apparently, in such cases, sequence data of DNA is not enough to distinguish them. Since taxonomists know that varying ploidy levels may affect morphology and gene flow, they often need to examine both the morphology and cytology of their study plants. But, if so, why should they limit their additional viewpoints for cariotype and DNA sequences other than classic morphology? We have nowadays a wide range of choices of strategies or viewpoints to analyze organisms, as I repeatedly mentioned in this article. Apparently, we are not allowed to limit our viewpoints for understanding of plants to morphology, chromosome number, and DNA sequences only, because we know that speciation of plants does not necessarily accompany changes in chromosome number or DNA sequence (see below). Let see researchers of Arabidopsis genetics again. When, we, molecular biologists analyze mutant of A. thaliana, we do not restrict our method to molecular techniques only. We use ALL available methods to understand the mutant plants. They are anatomy, cell biology, biochemistry, physiology, molecular biological analyses, transgenic systems, microarray analysis, ....etc., Why not taxonomists?
For example, we know that not only classic genetic mutation but also epigenetic mutations, in which sequence data of DNA or cariography should not detect any difference from their original strains, greatly affect the morphological features and process of evolution. A peloric type of Linaria vulgaris L. (Scrophulariaceae) was described by Linnaeus and has been thought to be a genetic variant of the standard type which has dorsoventrally asymmetric flowers. However, it was not a mutant in a classic sense. Cubas et al. (1999) found that the peloric type can be derived by an epigenetic mutation on the Linaria eye-like gene, Lcyc, that regulates floral symmetry. The peloric individuals of L. vulgaris have highly methylated Lcyc locus and the Lcyc is epi-genetically silenced. Methylation-dependent gene silencing is not stable and the peloric individuals sometimes reverts to develop wild-type flowers (Cubas et al., 1999). Since the Lcyc locus itself is not mutated at all in terms of the DNA sequence, the epi-mutant and wild-type individuals cannot be distinguished from DNA sequences, but can be distinguished by the expression level of Lcyc mRNA. Similar epigenetic mutations have been shown to be not rare. Moreover, we have accumulated vast proofs that an alteration of activity or function of a single gene can cause great effects on the morphology, physiology and total life of organisms, In fact, some cases of speciation which is tightly linked to morphological adaptations to a particular environment seem to be derived from a few sets of mutations (discussed in Tsukaya, 2002a). In some of such cases, molecular clock may be too rough to analyze the history of speciation, because morphological “jump”can cause rapid process of speciation which does not accumulate neutral mutations on DNA sequences enough to be detected, to date. Moreover, differed from neutral mutations, the above-mentioned mutations should be occurred on a few specific genes which are usually difficult to be identified from more than tens of thousands of genes of a plant genome, in particular, for wild, non-model species.
Microarray profiling of mRNA or transcriptome analysis may solve some of the problem and is expected to detect any differences on the level of accumulation of mRNA between the mother type and its derivatives. Microarray technology now allows us to analyze relative levels of expression (transcription) of huge number of genes at once. DNA molecules representing each gene are placed in a discrete spot on a base (usually a slide grass) at high density (more than four hundreds of spots per cm2). This is called a microarray (DNA chip). All genes of an organism which has relatively small size of genome, such as human and A. thaliana can be covered by a few microarrays. The messenger RNA to be analyzed are labeled with a fluorescent dye, and hybridized to the DNA dots on the microarray. Quantitative measurement of degree of hybridization on each spot is carried out by measuring the intensity of fluorescence from each spot after wash. This technology is basically designed for analysis of model organisms which all genome sequence was identified, because we need information on all possible genes in the genome to spot them on microarrays. A. thaliana and rice are the cases of plants. But it may be also possible to roughly analyze profile of mRNA of a non-model species from which genomic information has not yet obtained if we carry out cross-species microarray profiling with microarrays of A. thaliana or rice. Cross-species microarray means analysis of mRNA of a particular organisms with microarrays of its related model species. Cross-species microarray profiling is still rare but has begun to be tried to be applied in varied species (Moody et al., 2002; Becher et al., 2004;Weber et al., 2004). For example, analysis of pig mRNA by human microarrays or analysis of Arabidopsis halleri mRNA with microarrays of A. thaliana were successfully carried out. In the near future, cross-species microarray analysis will be applied also for taxonomic studies of wild plants. It should, I suppose, valid to be carried out to make a great leap forward in the plant taxonomy. But unfortunately, RNA is far more labile than DNA, and cannot be stored for a long time unless stored in deep freezer (at -80°C or so). Moreover, profile of transcribed mRNA is greatly affected by the physiological conditions and the environments, we cannot compare RNA profiles of two individuals which have separately grown in two distant areas. Therefore, correction of RNA in a field is inadequate and we should cultivate the plants to be investigated under the same growth conditions. Considering the importance of living materials not only for determination of chromosome number or RNA profiles but also for conservation of endangered species, botanical gardens in which various species of plants are cultivated are also required to collaborate with the above-mentioned collaborative activity among herbaria and DNA stock centers. Tight linkage among these three kinds of organizations, as a bioresource consortium, will greatly help studies of plants (Fig. 7).
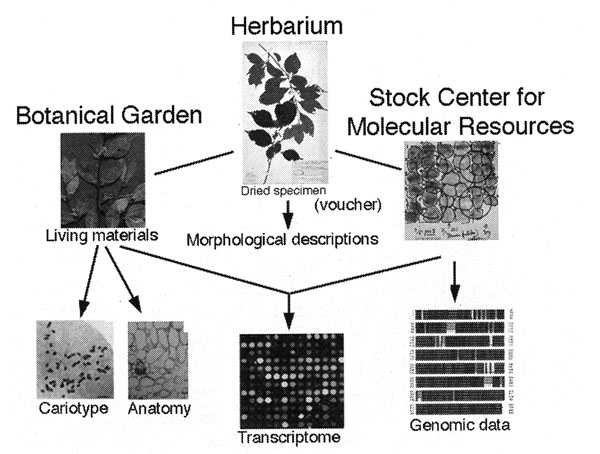
Fig. 7 An idea of consortium among herbaria, botanical gardens and stock center for molecular resources. Herbarium specimens are the basis of all activities in this consortium as vouchers. Cooperative activities among these organizations and worldwide researchers of plants may be linked via websites. See text for details.
Then, what is next? Considering that gene expression is regulated not only in the level of transcription but also in the post-transcription level, namely, translation of mRNA into a peptide, folding of peptides, modification of peptides, and so on, proteome profiling may be used for the next stage of analysis of plant taxa. Although viewpoints of some plant taxonomists are still engaged in the limited information which can be deduced from classic herbarium specimens that are not designed for some analytical studies, scientists should be free from tradition in pursuing truths. Renovating the status of specimens by tight collaboration of herbaria with stock centers of molecular resources and botanical gardens, I suppose, plant specimens will re-appear to be the most important basis of all studies of plants.